
Novel Drug Treatment Developed for Cystic Fibrosis
In October 2019, the Federal Drug Administration (FDA) approved a groundbreaking therapy against cystic fibrosis, a hereditary disease that gradually shuts down the lungs and pancreas. Researchers conducting an international clinical trial, which included patients and faculty of the University of Texas Southwestern Medical Center, found that administration of a cocktail drug, one combining elexacaftor, tezacaftor, and ivacafto, was highly effective in neutralizing symptoms associated with the disease.
More than 90% of patients afflicted with cystic fibrosis possess the Phe508del cystic fibrosis transmembrane conductance regulator (CFTR) allele, a genetic mutation that blocks the synthesis of the CFTR protein. The CFTR protein is involved in the regulation of mucus, sweat, saliva, tear, and digestive enzyme production. Without it, the body makes a dangerous excess of mucus, which can obstruct the pancreas and clog up the lungs. This mucus buildup can lead to long-term and potentially fatal health complications such as liver disease and diabetes.
Using a series of drugs that specifically addresses this genetic deficiency, the study found that patients receiving the experimental treatment over a 24 week period displayed significant improvement in lung function compared to the placebo group. Patients also exhibited a substantial reduction in lung flare-ups, which are episodes characterized by abnormal wheezing, weight loss, pneumonia, and sinusitis.
A further indication of the trial’s success was the patients’ decrease in “salty sweat,” a form of electrolyte loss manifesting in salt leakage via excess perspiration. Since cystic fibrosis affects the lungs and pancreas, patients are unable to efficiently digest foods and other nutrients. As a result, people with cystic fibrosis tend to lose more sodium and chloride salts, which can lead to a dangerously low blood pressure levels.
More than 90% of patients afflicted with cystic fibrosis possess the Phe508del cystic fibrosis transmembrane conductance regulator (CFTR) allele, a genetic mutation that blocks the synthesis of the CFTR protein. The CFTR protein is involved in the regulation of mucus, sweat, saliva, tear, and digestive enzyme production. Without it, the body makes a dangerous excess of mucus, which can obstruct the pancreas and clog up the lungs. This mucus buildup can lead to long-term and potentially fatal health complications such as liver disease and diabetes.
Using a series of drugs that specifically addresses this genetic deficiency, the study found that patients receiving the experimental treatment over a 24 week period displayed significant improvement in lung function compared to the placebo group. Patients also exhibited a substantial reduction in lung flare-ups, which are episodes characterized by abnormal wheezing, weight loss, pneumonia, and sinusitis.
A further indication of the trial’s success was the patients’ decrease in “salty sweat,” a form of electrolyte loss manifesting in salt leakage via excess perspiration. Since cystic fibrosis affects the lungs and pancreas, patients are unable to efficiently digest foods and other nutrients. As a result, people with cystic fibrosis tend to lose more sodium and chloride salts, which can lead to a dangerously low blood pressure levels.
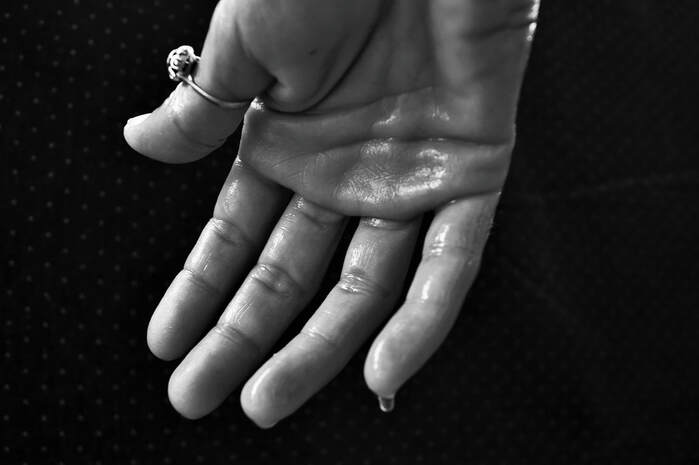
Excessive sweating dramatically reduces the body's salt reserves, and is a hallmark of cystic fibrosis.
Image Source: BarbaraBonanno
In this study, however, the treatment group was found to have lower concentrations of salts in their sweat. The drugs administered to this group all tackle different aspects of cystic fibrosis. While elexacaftor and ivacaftor stimulate the synthesis of the CFTR protein, thereby correcting its deficiency, tezacaftor opens more chloride channels in cells, increasing the uptake of salt in the body and preventing it from being “sweated” out. The fact that patients in the treatment group were observed to retain more salt demonstrates that that the elexacaftor–tezacaftor–ivacaftor combination is targeting the central mechanisms driving cystic fibrosis.
While this research serves as a potential game-changer for cystic fibrosis treatment, clinical research for the condition is ongoing. Innovative methods are needed for the remaining 10% of cystic fibrosis patients who lack the CFTR mutation, leaving them immune to this drug cocktail. Additionally, only time will tell if this treatment will adequately curb the symptoms of cystic fibrosis without any adverse side effects. However, people with the disease now have one more comprehensive medical opportunity to alleviate their most aggravating symptoms, and the study itself potentially paves the way for a cure that could benefit future generations to come.
While this research serves as a potential game-changer for cystic fibrosis treatment, clinical research for the condition is ongoing. Innovative methods are needed for the remaining 10% of cystic fibrosis patients who lack the CFTR mutation, leaving them immune to this drug cocktail. Additionally, only time will tell if this treatment will adequately curb the symptoms of cystic fibrosis without any adverse side effects. However, people with the disease now have one more comprehensive medical opportunity to alleviate their most aggravating symptoms, and the study itself potentially paves the way for a cure that could benefit future generations to come.
Featured Image Source: kalhh
RELATED ARTICLES
|
Vertical Divider

|
Vertical Divider
|
Vertical Divider
|
